Doug's DNA Page Continued
STRs and Haplotypes
Haplotypes determine the fine branches of the tree of Man, on the scale of families and hundreds of years, just as haplogroups determine the larger branches, on the scale of populations and tens of thousands of years. Haplotypes are defined by "STR"s. STR stands for "Short Tandem Repeat", also known as microsatellites. STRs are a very different type of mutation from SNPs and are distantly related to insertions or deletions, though they occur by a very different molecular mechanism. There are areas of DNA along the chromosome, with no known purpose, which consist of several repeated copies of some short (2 to 6 bases) motif. For example, the list of bases in the region known as DYS391 might be
TGTCTGTCTATCTATCTATCTATCTATCTATCTATCTATCTATCTATCTGCCT
which has ten copies of the motif TCTA flanked by different patterns. Sometimes the genetic copy mechanism "slips up" and copies the wrong number of repeats. This constitutes an STR mutation. The main measurement made on people in our DNA project consists of the measurement of the number of these repeats for a large number of "markers". DYS391 is one such marker. Others are given different names, many beginning with the designation DYS, others with random sounding names or names telling the repeat unit (like GATA-A10). The "DYS" is often left off of the designation of marker names, leaving only a number. There is also a type known as DYF, which is always listed, and note carefully that DYS385 and DYF385 are very different markers. Only a few Clan Donald men have yet tested DYF385. The numbers in our charts are just these repeat counts.
The list of all the numbers is called a haplotype. The numbers themselves really mean nothing to genealogy, only differences between people matter. Each marker mutates independently of all the others (but see below for DYS389). Using these mutations we can calculate how long ago two people shared a common ancestor. We can also, to some extent, determine whole genealogies from haplotypes (see Network Tree Charts below).
The marker DYS389 represents a special case in interpretion of our results. Two numbers are listed for this marker, called 389-1 and 389-2. There are two places on the Y chromosome where this marker occurs. The testing companies have devised two ways of testing their length. One way measures the length of just one, while the other way measures the sum of the lengths.The 389-1 listing is the single one and the 389-2 listing is the sum. The 389-1 number gives the length of one marker, and the difference between 389-2 and 389-1 gives the other length. Hence if one person is listed as having numbers 13 and 29 for 389-1 and 389-2 respectively the lengths are 13 and 16. If another person is listed at 14 and 30, the real numbers to compare are 14 and 16, so there really is only one mutation, not two.
Some other markers, DYS385, DYS459, DYS464, DYF371, DYF385, DYF397, DYF399, DYF401, DYF408, DYF411, and YCAII, all having two or more values, are prone to an entirely different type of mutation called a recLOH. This causes one of the copies to become a duplicate of the other. Thus 8-10 at 459 could become 10-10 or 8-8 in one event, not two separate ones. At DYS 464, 13-14-16-18 could become 16-16-18-18 in one event, not seven. You have to look for these yourself, as our software does not detect them.
TMRCA
By comparing haplotypes we can make rough estimates of how many generations have elapsed since two men shared a common male line ancestor. This is called the TMRCA (Time since Most Recent Common Ancestor). If one tests the 37 FTDNA markers, one expects to see one mutation every 7 generations. It is of great importance to realize that mutations happen at random, like throws of dice, rather than like a precise clock. One family line might actually have 3 mutations in ten generations (assuming one son per generation) while another might have none at all for 20 generations.
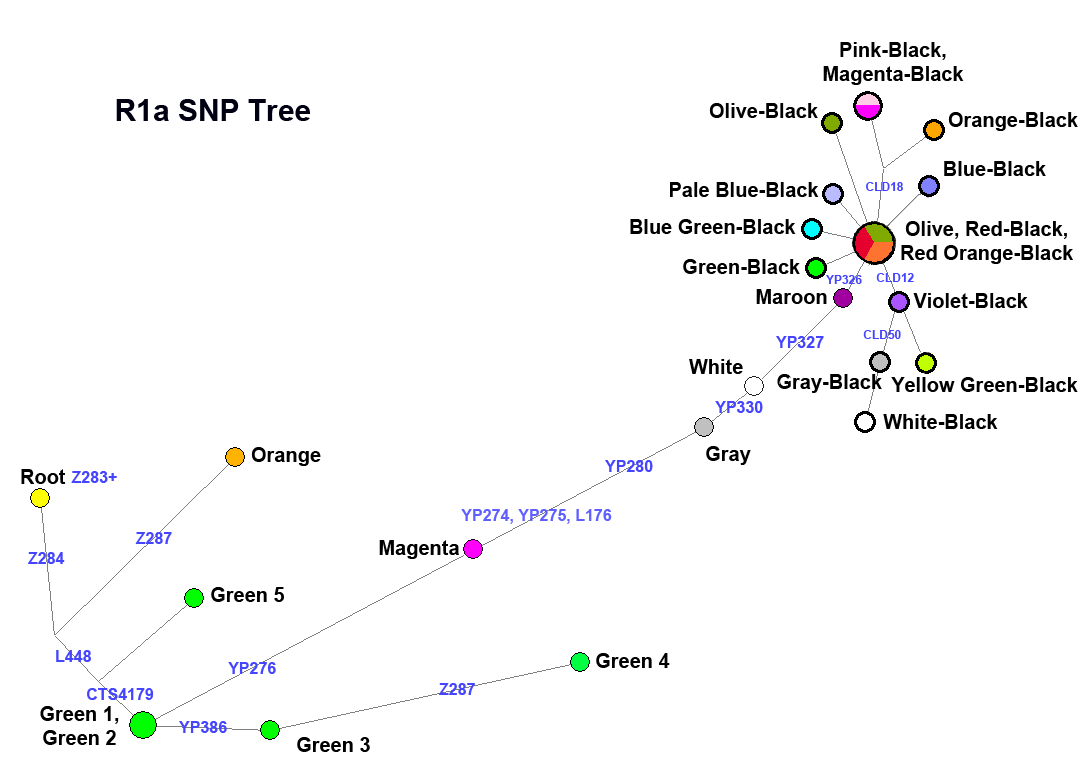
On average, men differing by one marker in 37 will have a common ancestor 4 back (roughly half of the 7 mentioned above, since either line could mutate). Our data charts give the number of mutations measured and this TMRCA. This number is actually just a rough mathematical estimate: half the time the real number will be greater, half the time it will be less. Sometimes it will be way off. 10% of the time it would be in fact 2 generations or less, and 10% of the time it would be expected to be 12 generations or more. "Rough estimate" really does mean rough. The only way to get better estimates is to pay for more markers. The graph below shows what the probability is that two men who match at 36 out of 37 markers have their most recent common ancestor at a certain generation back.
Similar methods can be used to predict when a larger group of people had a common all male line ancestor, or at least when a very small group of related people started a large increase in population. Our results tables give such a result for each subgroup.
Our TMRCA results are in generations since the MRCA. To get to years, you have to know how long a generation is. A good average number for Scotland before 1800 and the USA since 1607 is 31 years. There are several online TMRCA calculators, such as this one provided by us using our mutation rates.
Each STR on average mutates at a differing rate from other STRs. Using the Sorenson Foundation and Ymatch databases, plus several academic studies, as well as our own data on Somerled descendants, your webmaster has been able to infer the 'concensus' absolute mutation speeds of the Family Tree DNA 111-marker STR panel. These rates are needed for the various calculations. Note that Professor McDonald's rates are quite a bit slower than those used by FTDNA for their on-line TMRCA "FTDNATiP". We believe that ours are more reliable than those of FTDNA since TMRCA calculations with them agree perfectly with the same calculations using SNPs derived from BigY results.
Network Tree Charts
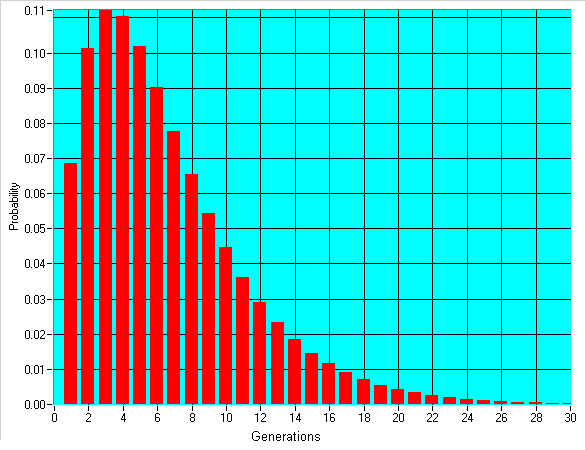
It is possible to infer relationships from DNA data, without starting with a paper trail. With the number of STR markers we have, even if everyone had 111 of them, it is not possible to do so with certainty. Doing that requires that we have SNP data. This is now availabe for some of our participants from the BigY or "SNP Panels". We use computer methods to process our data employing both SNPs (which hav higher prioroty) and STRs. We now have classification groups of varyibng reliability depending on the number of SNP tests a person has taken and the uniqueness of their STR results. We present the computer results for a part of R1a using our most stringent classification criteria.
These charts illustrate the genealogical "tree" of haplogroups. The path length is proportional to the number of mutations along it. These positions of branches on these charts are determined entirely on SNPs.
In some cases too few people have tested some "far out tree tip" SNPs to make colored groups for them. This is indicated on the the trees by multiple circles with the sames color. In other cases non-SNP data (i.e. surname or truly unambiguous STR data) allows assigning people with STR data. Since these trees use only the SNP data to position branches, there are pies with multiple colors, which represent these people.
We have labeled almost all major SNPs on the R1a chart, shown below. The circle with Olive, Red, and Red Orange is the location of Somerled. The blue labels are the names of the SNPs for major branches. This chart is seriously out of date, but illustrates this nicely. To see a current representation of data for both R1a and R1b, go to our BigY charts page .